Proceedings
Precision Pathology of Lymphoid Neoplasms
Mariusz Wasik,1 Deniz Peker Barclift,2 Xianfeng Frank Zhao3
1Department of Pathology, Fox Chase Cancer Center, Philadelphia, PA; 2Department of Pathology and Laboratory Medicine, Emory University, Atlanta, GA; 3Department of Pathology, University of Arizona College of Medicine Phoenix, Phoenix, AZ
Abstract
This section is dedicated to the precision pathology of lymphoid neoplasms. Dr. Mariusz Wasik in his keynote presentation, NPM::ALK: The prime example of potent oncogene and therapeutic target, discussed the critical role ALK1 plays in the pathogenesis of anaplastic large T-cell lymphoma (ALCL), why it serves as an optimal therapeutic target, and how to overcome the drug resistance. Dr. Deniz Peker Barclift, from the pathological diagnosis to detection of measurable residual disease, presented the molecular applications in the diagnosis and precision therapy of chronic lymphocytic leukemia (CLL). Dr. Xianfeng F. Zhao finally reviewed the recent molecular advances in diffuse large B-cell lymphoma (DLBCL) and shared his perspective on the precision pathology of DLBCL. These presentations together illustrate an exemplary landscape of precision pathology on both indolent and aggressive malignant lymphomas.
Keywords: ALCL, ALK1, CLL, DLBCL, NPM, precision pathology
Correspondence: Mariusz Wasik, MD, Department of Pathology, Fox Chase Cancer Center, Philadelphia, PA. Marriusz.Wasik@fccc.edu; Deniz Peker Barclift, MD, Department of Pathology and Laboratory Medicine, Emory University, Atlanta, GA. deniz.peker@emoryhealthcare.org. Xianfeng Frank Zhao, MD PhD, Department of Pathology, University of Arizona College of Medicine Phoenix, Phoenix, AZ. xfzhao@arizona.edu.
NPM::ALK: The Prime Example of Potent Oncogene and Therapeutic Target
ALK1 gene itself and its constitutively active oncogenic gene-fusion form NPM::ALK, were first identified in anaplastic large cell lymphoma (ALCL) of CD4+ T-cell origin [1]. As a member of tyrosine kinase family, ALK1 is a cell-membrane receptor physiologically expressed only in fetal neural cells, and it is important for differentiation and survival of these cells [2]. ALK1 is related to LTK and ROS, and remotely to insulin receptor. Follow-up studies identified a number of different ALK1 gene translocations with diverse partners in the whole spectrum of malignancies and in neuroblastoma, hence a tumor closely resembling fetal neural cells, full length of ALK1 can also acquire activating point mutations in the kinase domain (Table 1). Accordingly, in addition to ALCL, aberrant ALK1 expression has been detected in multiple malignancies which include B-cell lymphomas, inflammatory myofibroblastic tumor (IMFT), non-small cell lung carcinoma (NSCLC), renal medullary carcinoma (sickle cell trait-associated), anaplastic thyroid carcinoma, and neuroblastoma (sporadic and familial).
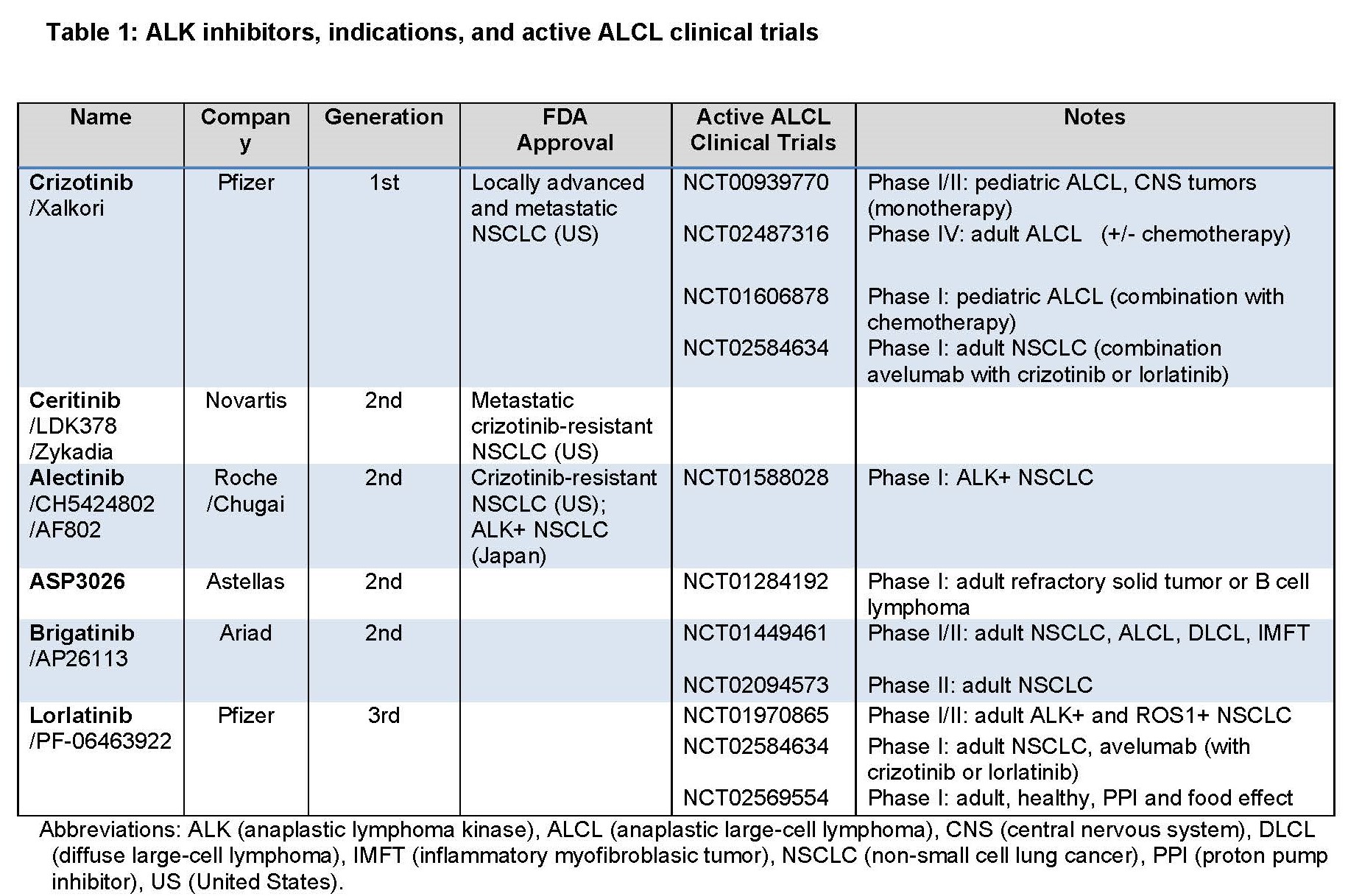
In regard to the underlying mechanism of NPM-ALK oncogenicity, ALK1 becomes aberrantly and constitutively expressed due to persistent activation of the NPM promoter in the host CD4+ T cells and the NPM-ALK protein is constitutively activated due to the NPM domain-mediated NPM-ALK oligomerization, resulting in the ALK kinase domain-triggered autophosphorylation [3]. NPM-ALK is highly oncogenic, both in vitro and in vivo in mouse models [3]. Experiments showed that induced expression of ALK1 results in malignant transformation of normal human CD4+ T lymphocytes; the cells became immortalized and larger in size [4]. These NPM-ALK transduced T cells are completely NPM-ALK-dependent, because their treatment with either ALK inhibitor or siRNA affects their size and inhibits their growth. In the xenografted animal models, the NPM-ALK transfected CD4+ T cells recapitulate the morphology and immunophenotype of human ALCL [4]. ALK+ ALCL patient studies have shown that of the 11 adult patients with advanced/therapy-resistant ALCL treated with a single agent, first-generation ALK inhibitor crizotinib, 7 achieved complete remission (CR) with 3 undergoing follow-up allotransplant consolidation [5,6]. Discontinuation of ALK inhibitor therapy in the responders resulted in disease recurrence [7]. In the pediatric patients treated with ALK inhibitors, 7 out of 9 patients achieved CR; some with just one cycle of treatment, with 3 undergoing autologous bone marrow transplant. In the follow up study of 26 pediatric/young adult patients, >80% patients achieved CR [8,9]. Due to the high efficacy, FDA approved crizotinib for ALK+ ALCL therapy in January 2021. Unfortunately, drug resistance frequently develops, mainly through ALK mutations [9]. New generations of ALK1 inhibitors have been produced and sequential treatment may be able to overcome the newly emerging drug-resistant mutations (Table 2) [10]. However, the drug resistance remains a challenge and multiagent novel combination therapy may well prove a strategy necessary to prevent the occurrence of drug resistance.
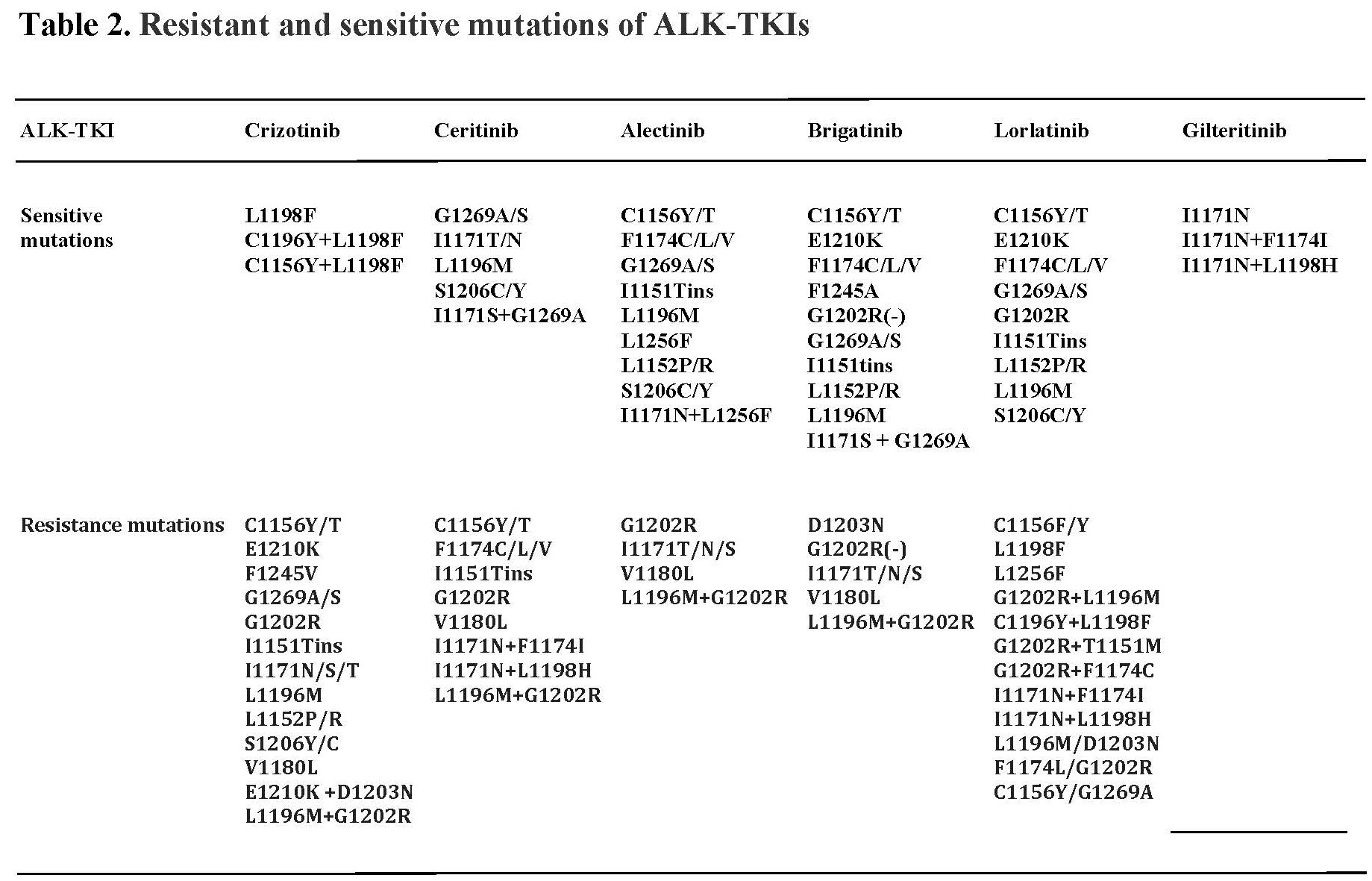
NPM-ALK activates several downstream signal transduction pathways [11], which include PI3K-AKT-FOXO3A/p27, mTOR, MEK-ERK, and JAK-STAT. NPM-ALK hijacks the preexisting signaling pathways, physiologically activated by IL2-type cytokines to promote the development and growth of ALCL [11]. Interestingly, ALK-negative ALCL uses a similar mechanism by activating ROS1, TYK2, and mutated JAK1 to eventually activate STAT3 in the transformation of normal T-cells [12,13]. Recent study showed that JAK2 translocations also play a role in activating the downstream cytokine pathways in the ALK-negative ALCL [14]. Through inducing epigenetic silencing of SHIP-1, STAT5A, and IL-2Rg, NPM-ALK protects its own constitutive expression [15-17], while it also promotes expression of DNMT1, the DNA-modifying enzyme needed to silence these genes [18]. By inhibiting IL2-type and T-cell receptor (TCR) signaling, NPM-ALK mediates the reprogramming of CD4+ T cells from TCR and IL-2-type signaling dependence to NPM-ALK dependence. ALK+ ALCL cells are protected from immune response by both secreting tolerogenic cytokines (IL-10 & TGF b) and expressing immunosuppressive cell-membrane protein CD274 (PD-L1) [19,20]. Of note, a chemotherapy- and ALK inhibitor-resistant ALK+ ALCL patient responded to anti-PD1 antibody and achieved complete remission [21], identifying potential novel therapy for ALK+ALCL and, perhaps, other ALK-driven malignancies. Our study also showed that NPM-ALK induces expression of ICOS gene: “parasitic” utilization of stimulatory signals generated by immune cells [22] and expression of HIF1α mRNA to protect malignant cells from effects of hypoxia [23]. It also induces expression of genes encoding stem-cell type transcription factors: TWIST1, SOX2, and SALL4 to confer the CD4+ T-cell features of immaturity [24-27], as well as the loss of T-cell identity [28]. NPM-ALK induces protection of lymphoma cells from apoptotic death via STAT3-HES1-TXNIP network [29].
In summary, NPM-ALK transforms CD4+ T cells by 1) constantly activating IL-2-type signaling pathways; 2) regulating key cell functions that include cell growth and hypoxia tolerance via HIF-1α, 3) modulating immune system (IL-10, TGFβ, CD274/PD-L1, and ICOS) to suppress anti-tumor response; 4) affecting epigenetics via STAT3-promoted expression of DNMT1 by inducing transcription of DNMT1 gene and by suppressing expression of its translational inhibitor miR-21; 5) inducing via STAT3 epigenetic silencing of tumor suppressor genes SHP-1, STAT5A, and IL-2Rγ to protect its own expression; 6) inducing via STAT3 expression of HES1 to inhibit the expression of pro-apoptotic tumor suppressor TXNIP, and enhancing cell metabolism by activating mTORC1. Therefore, NPM-ALK is an omnipotent oncogene and serves as an optimal target for precision cancer therapy.
Molecular Applications in Chronic Lymphocytic Leukemia (CLL): From Diagnosis to Detection of Measurable Residual Disease
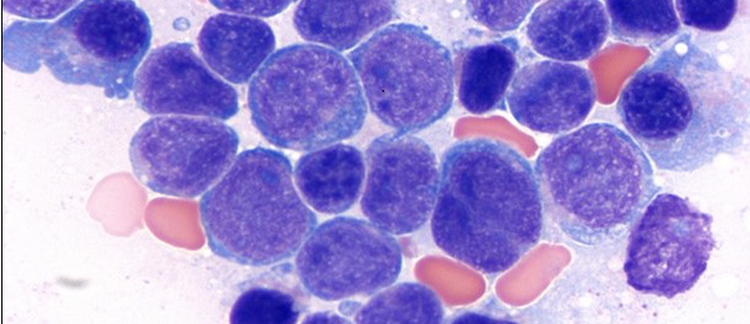
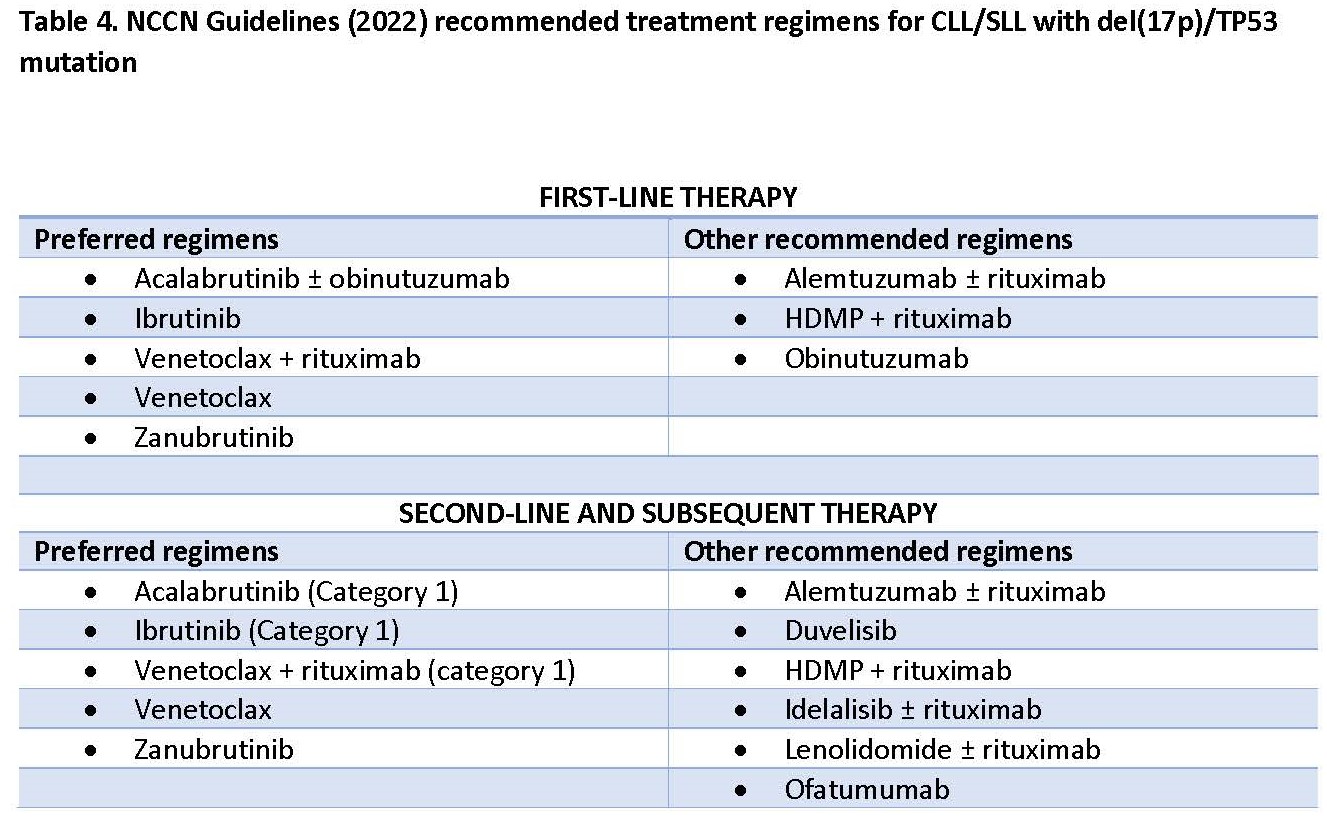
Chronic lymphocytic leukemia/small lymphocytic lymphoma (CLL/SLL) is an indolent neoplasm of mature B-cells. CLL is the most frequent leukemia in Western countries with 5 cases per 100,000 people. It is more common in Caucasians and has a slight male predilection. Familial predisposition exists with 2- to 8-fold increased risk in first degree relatives. CLL often has a stable course with risk of an aggressive disease and transformation to diffuse large B-cell lymphoma (Richter transformation). Diagnosis of CLL requires a comprehensive full body work up including blood and bone marrow examination using histomorphology, flow cytometry, molecular testing and body imaging to determine the disease extension. For diagnosis of CLL, monoclonal B-cells should be equal to or more than 5 x 109/L in blood or, if less than 5 x 109/L, should be accompanied by organomegaly and/or extramedullary disease. Blood and bone marrow show lymphocytosis with small, mature lymphocytes displaying condensed chromatin with “soccer ball” appearance and scant cytoplasm. Admixed few prolymphocytes can be observed and should not be in increased numbers. Flow cytometric analysis displays a typical immunophenotype with expression of CD5, CD23 and CD200 in B-cells in most cases. ZAP70 and CD38 expression can be determined by flow cytometry and are used as prognostic indicators in CLL as they reflect IGHV mutation status. LEF1 immunohistochemical stain has been used to determine CLL/SLL origin and cyclin D1 and SOX11 should not be expressed. Molecular testing in CLL is important, particularly at the time of first diagnosis and should include cytogenetics, FISH and, preferably, mutation analysis for TP53 gene (please see Table 3 for frequent cytogenetic abnormalities in CLL and prognostic implications). Results of these testing are used in CLL risk stratification in the clinical setting.

IGHV mutation status is a useful prognostic parameter and potentially influences the type of therapy, i.e. immunotherapy vs. novel agent-based therapy. However, since the mutational status considered to be stable over the course of the disease, serial testing is generally not required. Next generation sequencing (NGS) has been used in CLL; however, it is still not a widely used diagnostic test. In addition to TP53 gene, SF3B1, ATM, NOTCH1, and BIRC3 genes were shown to be important prognostic indicators in CLL [30], particularly in determining effective treatment. Approximately 8% of CLL patients have TP53 alterations in the form of TP53 deletion, TP53 mutation, or harboring both, and are associated with inferior clinical outcome. It is crucial to determine the status of this gene as chemoimmunotherapy is not recommended in these patients and other regimens should be considered as recommended by NCCN Guidelines 2022 (Table 4). Recent studies have shown that SF3B1 and BIRC3 mutations, like TP53, are associated with inferior clinical outcome and fludarabine refractory disease [31]. Emerging data from a clinical trial indicate that targeted therapeutic agents may be considered in these patients, such as Bruton tyrosine kinase (BTK) inhibitors and BCL2 inhibitor venetoclax [32].
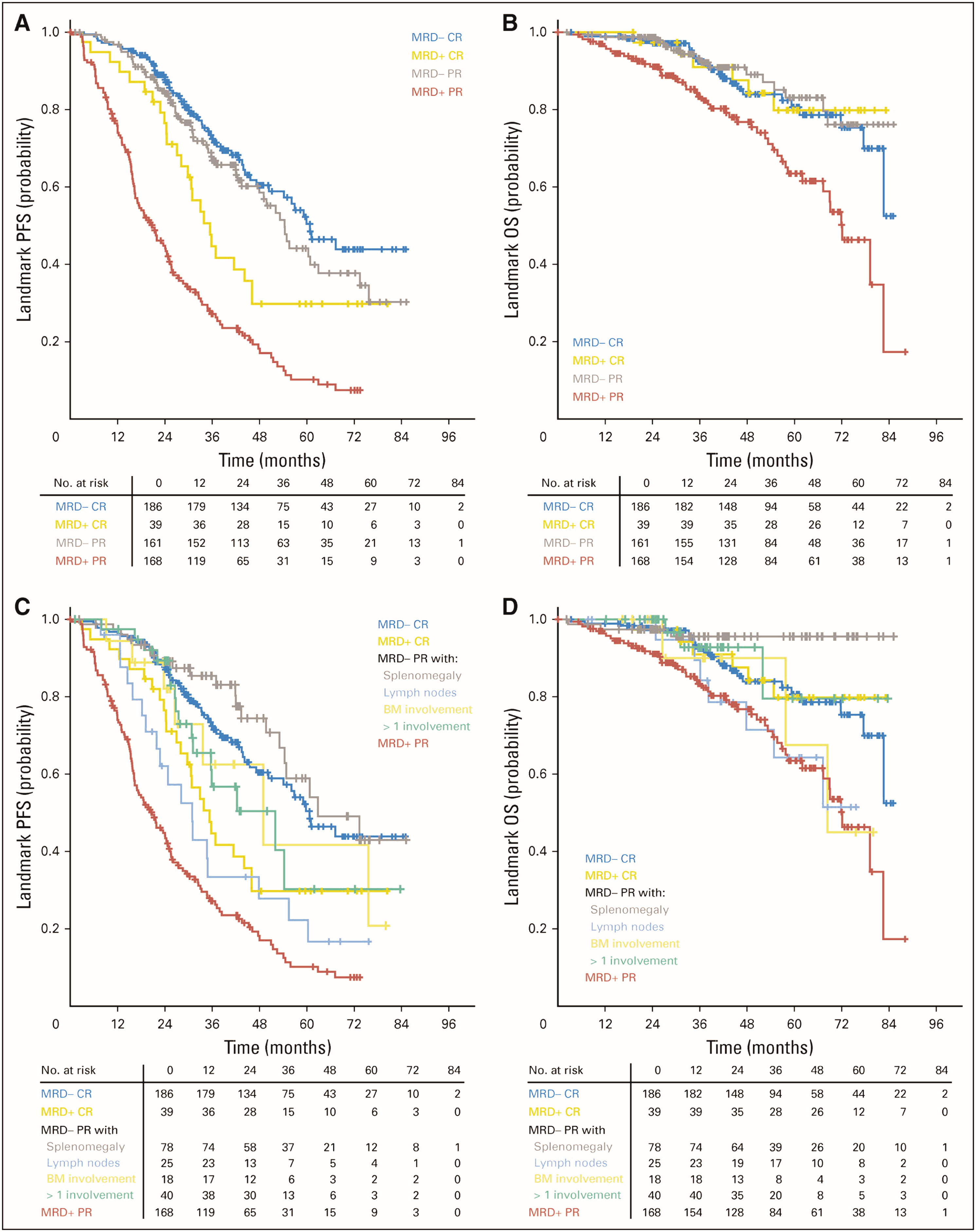
Minimal measurable or residual disease (MRD) is an emerging parameter to determine treatment response in CLL patients and current data [33] suggest that partial clinical response with undetectable MRD might have superior overall prognosis when compared to complete clinical response with MRD (Figure 1). Therefore, more effective regimens to achieve complete remission without detectible MRD have been investigated. Results of a recent clinical trial showed promising results with combination of acalabrutinib, venetoclax, and obinutuzumab as frontline treatment to reach complete remission without detectable MRD in CLL patients regardless of TP53 and IGHV mutation status [34]. Currently, flow cytometry is the most widely used technique to measure MRD with alternative testing including allele-specific oligonucleotide PCR and NGS.
Precision Pathology of Diffuse Large B-cell Lymphoma (DLBCL)
As the most common type of non-Hodgkin lymphoma in the United States, diffuse large B-cell lymphoma (DLBCL) is a heterogeneous group of lymphoid malignancies [35], and the treatment remains largely rituximab + (cyclophosphamide, hydroxydaunorubicin, vincristine sulfate, and prednisone) (RCHOP) with some variations. The current regimen and dosage are based on numerous clinical trials; unfortunately, only 60-70% respond to the regimen and 40-50% of patients relapse [36]. Therefore, novel therapies are badly needed for DLBCL, which include targeted therapy.
DLBCL has been classified through the years with its vividly descriptive name. Based on gene profiling data [37,38], WHO Classification currently divides DLBCL into three major subtypes: 1) activated B-cell like (ABC) subtype (with a 5-year survival rate of 35%); 2) germinal center B-cell like (GCB) subtype (with a 60% 5-year overall survival rate); and 3) unclassified subtype (with a 5-year survival rate of 39%). GCB subtype DLBCL is associated with perturbations in the PI3K/AKT and JAK/STAT signaling pathways and a higher frequency of BCL2, EZH2 and MYC mutations, whereas ABC subtype DLBCL is characterized by activation of the BCR-mediated NF-κB and PI3K/AKT pathways, and a higher frequency of BCL6 rearrangements, BCL2 amplifications, and recurrent mutations of CD79B, MYD88, and PRDM1 [39,40]. Using shared genetic abnormalities, DLBCL has recently been classified into four genetic subtypes [41]: 1) MCD subtype (based on the co-occurrence of MYD88L265P and CD79B mutations); 2) BN2 subtype (based on BCL6 fusions and NOTCH2 mutations); 3) N1 subtype (based on NOTCH1 mutations); and 4) EZB subtype (based on EZH2 mutations and BCL2 translocations).
The most significant advance in DLBCL targeted therapy is the ibrutinib effect on DLBCL [42]. Ibrutinib, a BTK inhibitor, in combination with RCHOP gained a 100% response rate in a subgroup of young patients and the response is highly dependent on the mutations fostering the B-cell receptor (BCR) signaling and BTK dependence [43]. It is not known if ibrutinib by itself would gain a similar response without RCHOP. Another study by the same team found that the sole agent ibrutinib inhibitory effect on ABC subtype DLBCL indeed depended on the activated BCR signaling rather than the MYD88 mutation. However, when the mutated MYD88 became independent of BCR activation, ibrutinib lost its inhibitory effect on the ABC subtype DLBCL, even with NF-kB pathway activation [44].
With increasing popularity of precision medicine, many studies are being performed on targeted agents for the relapsed and refractory DLBCL [45]. However, these studies have some issues. For example, since most of the studies are for either relapsed or refractory diseases, it is hard to evaluate the effects on primary DLBCL. Considering all the medical conditions, the studies did not include pharmacomics which is critical for precision medicine. Population-based dosages led to incorrect efficacy and toxicity in individualized patients. Most of the studies are combined with non-targeting agents [42,43,45] and the effects of the individual targeting agent are not well evaluated. Current clinical trials are still population-based, which could misinterpret the clinical outcomes in individual patients. Due to these issues, the current classification has been questioned by clinicians [46,47], and it seems that the traditional classification no longer meets the requirement of precision medicine.
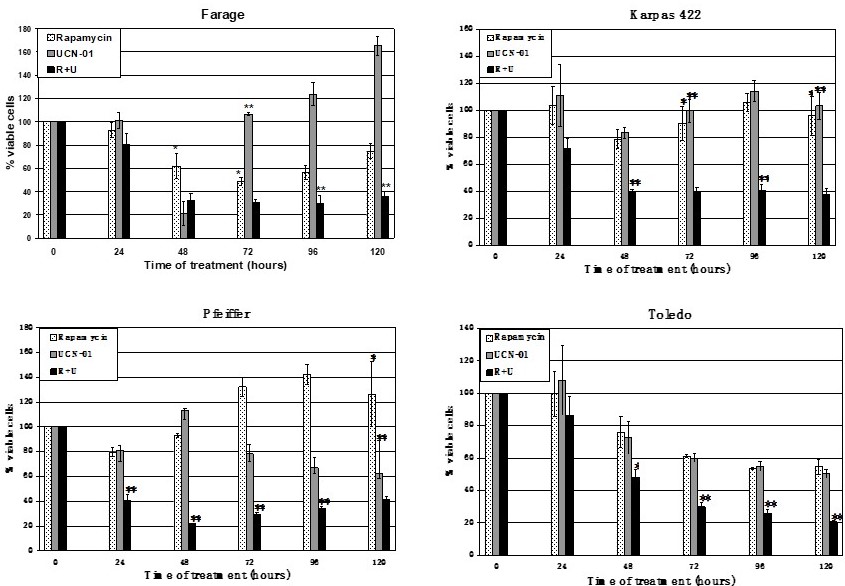
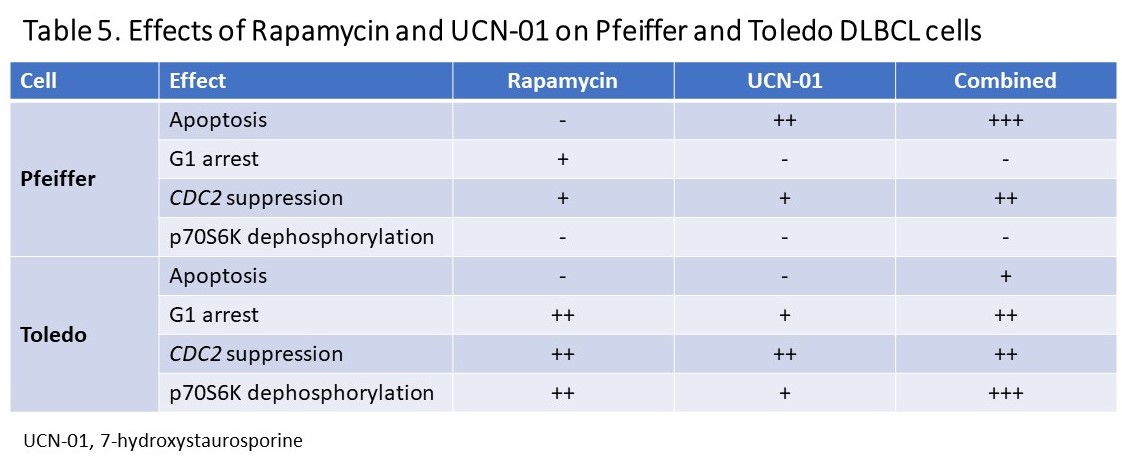
Previous study showed that treatment of lymphoma should be individualized [48]. In an investigation of DLBCL transformation, amplified RPS6KB1 and CDC2 genes were identified in the transformed large B-cell lymphoma by comparative genomic hybridization (CGH). Further study showed that the activated RPS6KB1 product, p70S6K, and CDC2 serine/threonine kinases were abundant in DLBCL specimens [48], and thus the activated (phosphorylated) p70S6K and CDC2 could be therapeutic targets for DLBCL. When four DLBCL cell lines were treated with rapamycin and/or UCN-01 for 5 days viable lymphoma cells were markedly decreased. Except for Toledo cells, the other 3 cell lines showed brief inhibition and subsequent recovery of cell proliferation with single agents. When combined, rapamycin and UCN-01 synergistically inhibited proliferation of both Pfeiffer and Toledo DLBCL cells (Figure 2). Analysis of cell cycle progression of the two DLBCL cell lines revealed that UCN-01 effect dominated in Pfeiffer cells, whereas G1 arrest dictated Toledo cells with predominant rapamycin effect (Table 5). Apoptosis played a major role in Pfeiffer cells but a minor role in Toledo cells. Although DLBCL cell proliferation were inhibited in both Pfeiffer and Toledo cells, they employed different mechanisms – in Pfeiffer cells, the dominant effect of UCN-01 caused apoptosis whereas in Toledo cells, rapamycin led to G1 arrest. Furthermore, rapamycin also inhibited the phosphorylation of p70S6K in Toledo cells, but not in Pfeiffer cells.
In addition to the PI3K/AKT/mTOR/p70S6K pathway, ERK1/2 and CHK2 genes were also overexpressed in DLBCL cells [49]. Either single agents or in combination, ERK1/2 and CHK2 inhibitors led the DLBCL cells to apoptosis. When the inhibitors combined, apoptosis was more prominent in both DLBCL cell lines and the xenografted tumors of the SCID mouse model. Crosstalk between the PI3K/AKT/mTOR and RAS/RAF/MEK/ERK pathways has been implicated in cancer pathogenesis and targeted therapies [50,51]. Therefore, multiagent combination therapy is likely an effective strategy to overcome drug resistance [52].
For precision diagnosis of DLBCL, excisional biopsy is preferred whenever possible since more tumor cells will be available for various studies. The specimen should be divided into at least three parts for: 1) morphology and immunophenotyping for pathological diagnosis; 2) NGS mutational analyses of the BCR, NF-κB, JAK/STAT, BCL2, BCL6, MYC, PI3K/AKT/mTOR/p70S6K, and RAS/RAF/MEK/ERK pathways and other critical components; 3) lymphoma cell culture for selecting therapeutic agents with optimal efficacy. Once the drugs are selected, draw patient’s blood for pharmacomics to determine the metabolizing rate, xenobiotic effects, drug-drug interactions, and adverse effects of the therapeutic drugs.
Therefore, the future pathology report should include: a) pathology diagnosis based on morphology and immunophenotype; b) NGS results for BCR, NF-κB, JAK/STAT, PI3K/AKT/mTOR/p70S6K, and RAS/RAF/MEK/ERK pathways and other critical components; c) activated pathway kinase profiles; d) drug efficacies on the lymphoma cells; e) pharmacomics of the patient; f) drug toxicity and other adverse effects on the patient.
Future clinical trials should be individualized which include screening the drugs that inhibit proliferation of the DLBCL cells with high efficacy and screening the patients with pharmacomics to identify the subjects who more likely will respond with minimal toxicity in the body. The other patients, even though with the same diagnosis, should not be included in the clinical trial.
In summary, precision pathology of DLBCL remains a challenge for pathologists. For the best clinical outcome of patients, we should use all the molecular data, including pathway and cell cycle kinase activities, to select the most efficacious drugs, and pharmacomics results to identify patients who benefit most from the selected therapeutic drugs. The “trial and error” clinical practice should be ended.
Acknowledgment
The authors claim no conflict of interest with the presentations.
References
- Morris SW, Kirstein MN, Valentine MB, et al. Fusion of a kinase gene, ALK, to a nucleolar protein gene, NPM, in non-Hodgkin's lymphoma. Science. 1994;263:1281-1284.
- Yao S, Cheng M, Zhang Q, Wasik M, Kelsh R, Winkler C. Anaplastic lymphoma kinase is required for neurogenesis in the developing central nervous system of zebrafish. PloS one. 2013;8:e63757.
- Werner MT, Zhao C, Zhang Q, Wasik MA. Nucleophosmin-anaplastic lymphoma kinase: the ultimate oncogene and therapeutic target. Blood. 2017;129:823-831.
- Zhang Q, Wei F, Wang HY, et al. The potent oncogene NPM-ALK mediates malignant transformation of normal human CD4(+) T lymphocytes. Am J Pathol. 2013;183(6): 1971-1980.
- Gambacorti-Passerini C, Messa C, Pogliani EM. Crizotinib in anaplastic large-cell lymphoma. N Engl J Med. 2011;364:775-776.
- Gambacorti-Passerini C, Farina F, Stasia A, et al. Crizotinib in advanced, chemoresistant anaplastic lymphoma kinase-positive lymphoma patients. J Natl Cancer Inst. 2014;106:djt378.
- Gambacorti-Passerini C, Mussolin L, Brugieres L. Abrupt relapse of ALK-positive lymphoma after discontinuation of Crizotinib. N Engl J Med. 2016;374:95-96.
- Mossé YP, Lim MS, Voss SD, et al. Safety and activity of crizotinib for paediatric patients with refractory solid tumours or anaplastic large-cell lymphoma: a Children’s Oncology Group phase 1 consortium study. Lancet Oncol. 2013;14:472-480.
- Mossé YP, Voss SD, Lim MS, et al. Targeting ALK with Crizotinib in pediatric anaplastic large cell lymphoma and inflammatory myofibroblastic tumor: A Children's Oncology Group Study. J Clin Oncol. 2017;35:3215-3221.
- Wang Y, He J, Xu M, et al. Holistic view of ALK TKI resistance in ALK-positive anaplastic large cell lymphoma. Front Oncol. 2022;12:815654.
- Marzec M, Halasa K, Liu X, et al. Malignant transformation of CD41 T lymphocytes mediated by oncogenic kinase NPM/ALK recapitulates IL-2-induced cell signaling and gene expression reprogramming. J Immunol. 2013;191:6200-6207.
- Lobello C, Tichy B, Bystry V, et al. STAT3 and TP53 mutations associate with poor prognosis in anaplastic large cell lymphoma. Leukemia. 2021;35:1500-1505.
- Crescenzo R, Abate F, Lasorsa E, et al. Convergent mutations and kinase fusions lead to oncogenic STAT3 activation in anaplastic large cell lymphoma. Cancer Cell. 2015;27(4):516-532.
- Fitzpatrick MJ, Massoth LR, Marcus C, et al. JAK2 rearrangements are a recurrent alteration in CD30+ systemic T-cell lymphomas with anaplastic morphology. Am J Surg Pathol. 2021;45:895-904.
- Zhang Q, Raghunath PN, Vonderheid E, Odum N, Wasik MA. Lack of phosphotyrosine phosphatase SHP-1 expression in malignant T-cell lymphoma cells results from methylation of the SHP-1 promoter. Am J Pathol. 2000;157:1137-1146.
- Zhang Q, Wang HY, Liu X, Wasik MA. STAT5A is epigenetically silenced by the tyrosine kinase NPM1-ALK and acts as a tumor suppressor by reciprocally inhibiting NPM1-ALK expression. Nat Med. 2007;13:1341-1348.
- Zhang Q, Wang HY, Woetmann A, Raghunath PN, Odum N, Wasik MA. STAT3 induces transcription of the DNA methyltransferase 1 gene (DNMT1) in malignant T lymphocytes. Blood. 2006;108:1058-1064.
- Zhang Q, Wang HY, Liu X, Bhutani G, Kantekure K, Wasik M. IL-2R common gamma-chain is epigenetically silenced by nucleophosphinanaplastic lymphoma kinase (NPM-ALK) and acts as a tumor suppressor by targeting NPM-ALK. Proc Natl Acad Sci USA. 2011;108:11977-11982.
- Kasprzycka M, Marzec M, Liu X, et al. Nucleophosmin/anaplastic lymphoma kinase NPM-ALK oncoprotein induces T regulatory cell phenotype by activating STAT3. Proc Natl Acad Sci USA. 2006;103:9964-9969.
- Marzec M, Zhang Q, Goradia A, et al. Oncogenic kinase NPM/ALK induces through STAT3 expression of immunosuppressive protein CD274 (PD-L1, B7-H1). Proc Natl Acad Sci USA. 2008;105:20852-20857.
- Hebart H, Lang P, Woessmann W. Nivolumab for refractory anaplastic large cell lymphoma: A case report. Ann Intern Med. 2016;165:607-608.
- Zhang Q, Wang H, Kantekure K, et al. Oncogenic tyrosine kinase NPM-ALK induces expression of the growth-promoting receptor ICOS. Blood. 2011;118:3062-3071.
- Marzec M, Liu X, Wong W, et al. Oncogenic kinase NPM/ALK induces expression of HIF1a mRNA. Oncogene. 2011;30:1372-1378.
- Zhang J, Wang P, Wu F, et al. Aberrant expression of the transcriptional factor Twist1 promotes invasiveness in ALK-positive anaplastic large cell lymphoma. Cell Signal. 2012;24:852-858.
- Gelebart P, Hegazy S, Wang P, et al. Aberrant expression and biological significance of Sox2, an embryonic stem cell transcriptional factor, in ALK-positive anaplastic large cell lymphoma. Blood Cancer J. 2012;2:e82.
- Wang P, Zhang JD, Wu F, et al. The expression and oncogenic effects of the embryonic stem cell marker SALL4 in ALK-positive anaplastic large cell lymphoma. Cell Signal. 2012;24:1955-1963.
- Lai R, Ingham RJ. The pathobiology of the oncogenic tyrosine kinase NPM-ALK: a brief update. Ther Adv Hematol. 2013;4:119-131.
- Pawlicki JM, Cookmeyer DL, Maseda D, et al. NPM/ALK-induced reprogramming of mature TCR-stimulated T cells results in dedifferentiation and malignant transformation. Cancer Res. 2021;81:3241-3254.
- Zhang Q, Wang HY, Nayak A, et al. Induction of transcriptional inhibitor HES1 and the related repression of tumor-suppressor TXNIP are important components of cell-transformation program imposed by oncogenic kinase NPM-ALK. Am J Pathol. In revision.
- Nadeu F, Delgado J, Royo C, et al. Clinical impact of clonal and subclonal TP53, SF3B1, BIRC3, NOTCH1, and ATM mutations in chronic lymphocytic leukemia. Blood. 2016;127:2122-2130.
- Rossi D, Bruscaggin A, Spina V, et al. Mutations of the SF3B1 splicing factor in chronic lymphocytic leukemia: association with progression and fludarabine-refractoriness. Blood. 2011;118:6904-6908.
- Tausch, E, Stilgenbauer S. BIRC3 mutations in chronic lymphocytic leukemia – uncommon and unfavorable. Haematologica. 2020;105:255-256.
- Wierda, W.G., Rawstron, A., Cymbalista, F. et al. Measurable residual disease in chronic lymphocytic leukemia: expert review and consensus recommendations. Leukemia. 2021; 35:3059-3072.
- Davids MS, Lampson BL, Tyekucheva S, et al. Acalabrutinib, venetoclax, and obinutuzumab as frontline treatment for chronic lymphocytic leukaemia: a single-arm, open-label, phase 2 study. Lancet Oncol. 2021;22:1391-1402.
- Gascoyne RD, Chan JKC, Campo E, et al. Diffuse large B-cell lymphoma, NOS. In Swerdlow SH, Campo E, Harris NL, Jaffe ES, Pileri SA, Stein H, Thiele J (Eds): WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues (Revised 4th Edition). IARC: Lyon 2017. p291-297.
- Crump M, Neelapu SS, Farooq U, et al. Outcomes in refractory diffuse large B-cell lymphoma: results from the international SCHOLAR-1 study. Blood. 2017;130:1800-1808.
- Alizadeh AA, Eisen MB, Davis RE, et al. Distinct types of diffuse large B-cell lymphoma identified by gene expression profiling. Nature. 2000;403:503-11.
- Rosenwald A, Wright G, Chan WC, et al. The use of molecular profiling to predict survival after chemotherapy for diffuse large-B-cell lymphoma. N Engl J Med. 2002;346:1937-1947.
- Abramson JS. Hitting back at lymphoma: How do modern diagnostics identify high‐risk diffuse large B‐cell lymphoma subsets and alter treatment? Cancer. 2019;125:3111-3120.
- Roschewski M, Staudt LM, Wilson WH. Diffuse large B-cell lymphoma-treatment approaches in the molecular era. Nature Rev Clin Oncol. 2014;11:12-23.
- Schmitz R, Wright GW, Huang DW, et al. Genetics and pathogenesis of diffuse large B-cell lymphoma. N Engl J Med. 2018;378:1396-1407.
- Younes A, Sehn LH, Johnson P, et al. Randomized Phase III Trial of Ibrutinib and Rituximab Plus Cyclophosphamide, Doxorubicin, Vincristine, and Prednisone in Non-Germinal Center B-Cell Diffuse Large B-Cell Lymphoma. J Clin Oncol. 2019;37:1285-1295.
- Wilson WH, Wright GW, Huang DW, et al. Effect of ibrutinib with R-CHOP chemotherapy in genetic subtypes of DLBCL. Cancer Cell. 2021;39:1643-1653.
- Wilson WH, Young RM, Schmitz R, et al. Targeting B cell receptor signaling with ibrutinib in diffuse large B cell lymphoma. Nature Med. 2015;21:922-926.
- Susanibar‐Adaniya S, Barta SK. 2021 Update on Diffuse large B cell lymphoma: A review of current data and potential applications on risk stratification and management. Am J Hematol. 2021;96:617-629.
- Nowakowski GS, Czuczman MS. ABC, GCB, and Double-Hit Diffuse Large B-Cell Lymphoma: Does Subtype Make a Difference in Therapy Selection? Am Soc Clin Oncol Educ Book. 2015:e449-457.
- Morin RD, Scott DW. DLBCL subclassification: divide and conquer? Blood. 2020;135:1722-1724.
- Zhao MY, Auerbach A, D'Costa AM, et al. Phospho-p70S6K/p85S6K and cdc2/cdk1 are novel targets for diffuse large B-cell lymphoma combination therapy. Clin Cancer Res. 2009;15:1708-1720.
- Dai B, Zhao XF, Mazan-Mamczarz K, et al. Functional and molecular interactions between ERK and CHK2 in diffuse large B-cell lymphoma. Nature Commun. 2011;2:1-9.
- De Luca A, Maiello MR, D'Alessio A, Pergameno M, Normanno N. The RAS/RAF/MEK/ERK and the PI3K/AKT signalling pathways: role in cancer pathogenesis and implications for therapeutic approaches. Expert Opin Ther Targets. 2012;16 Suppl 2:S17-27.
- Tasian SK, Teachey DT, Rheingold SR. Targeting the PI3K/mTOR pathway in pediatric hematologic malignancies. Front Oncol. 2014;4:1-8.
- Zhao XF, Gartenhaus R. Phospho-p70S6K and cdc2/cdk1 as therapeutic targets for diffuse large B-cell lymphoma. Expert Opin Ther Targets. 2009;13:1085-1093.